

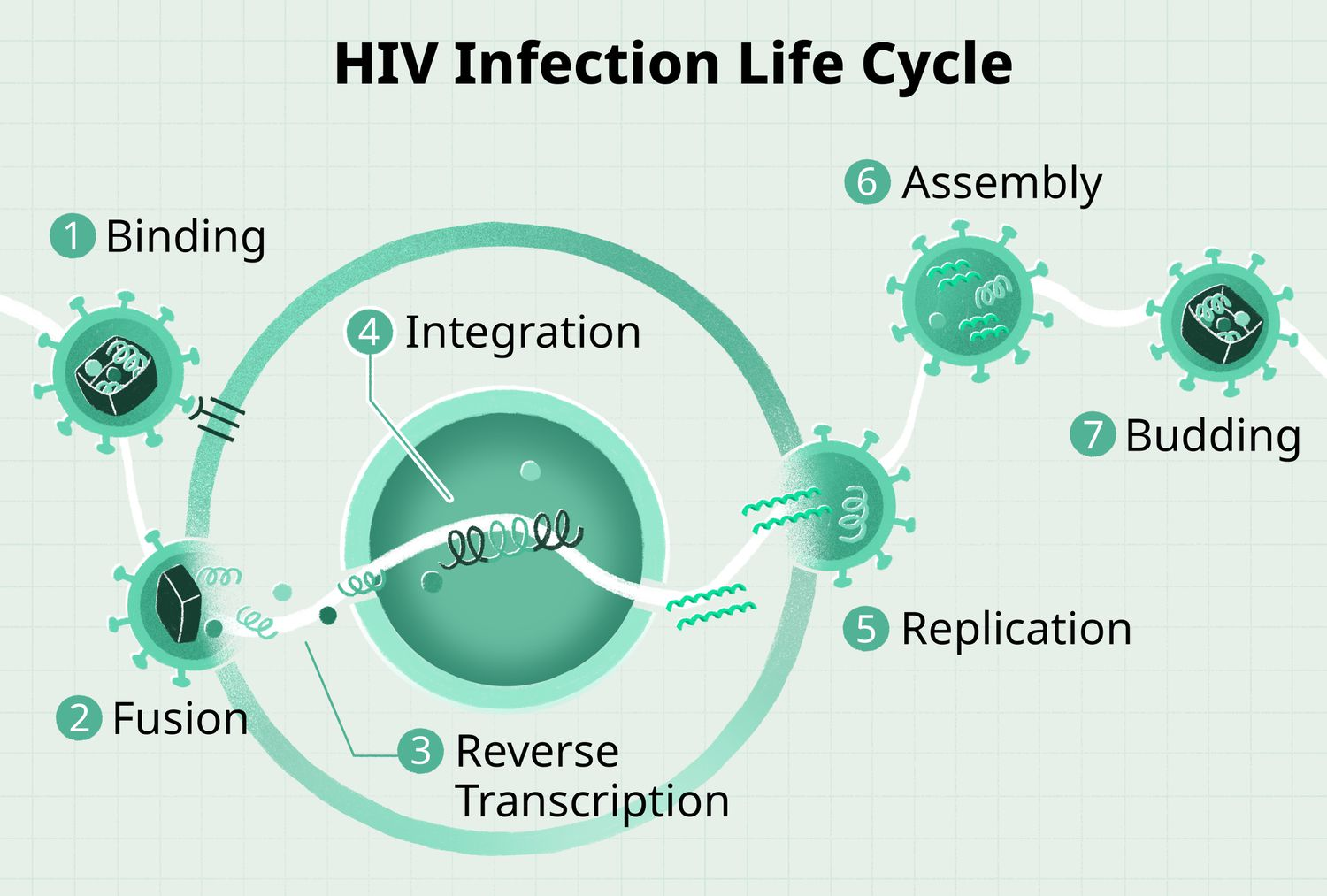
The HIV Virus Life Cycle involves distinct stages—from cellular binding and integration to replication and maturation. Modern Antiretroviral Therapy (ART) is strategically designed to target these specific viral mechanisms, helping suppress replication and overcome the evolutionary challenges of HIV drug resistance.
The HIV life cycle describes how the virus infects CD4 immune cells, replicates using the host’s machinery, and produces new infectious particles, typically completing one cycle in 1 to 2 days.
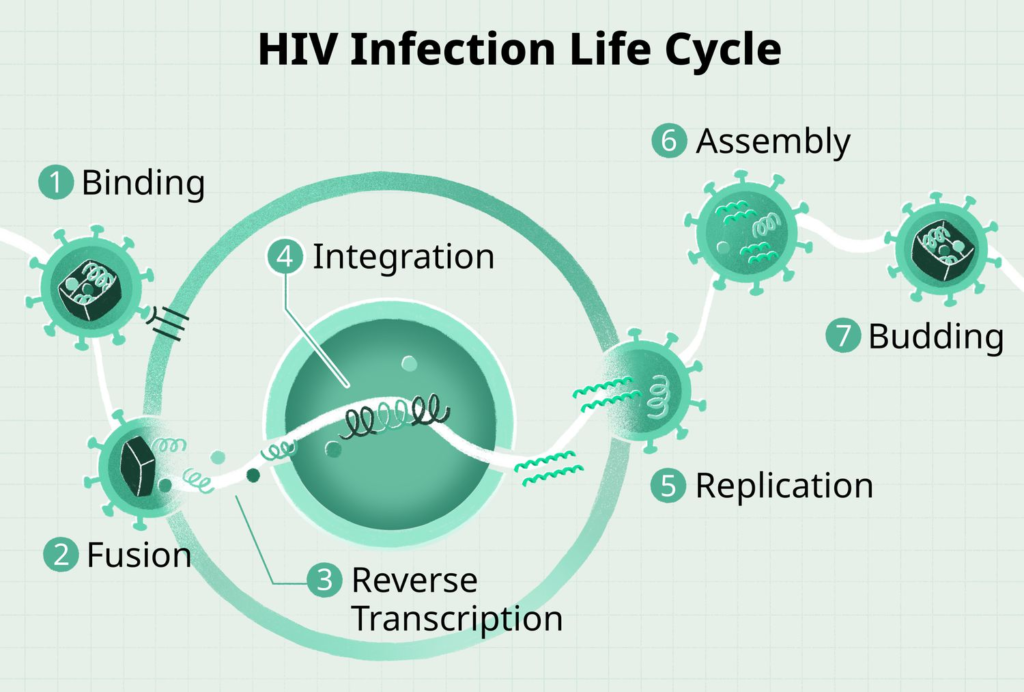
What Are the Key Stages of the HIV Virus Life Cycle?
HIV targets CD4 T-cells by attaching via gp120 glycoproteins to CD4 receptors and co-receptors (CCR5 or CXCR4), followed by fusion, where the viral envelope merges with the cell membrane to release RNA and enzymes inside. Reverse transcription converts viral RNA into DNA using reverse transcriptase; this DNA then enters the nucleus.
How Does HIV Integrate and Replicate Inside the Cell?
Integrase enzymes insert the viral DNA into the host cell’s genome, allowing transcription into viral mRNA and translation into proteins. New viral RNA and proteins assemble at the cell membrane, bud out as immature virions, and mature via protease cleavage into infectious particles ready to infect more cells.
Why is the HIV Life Cycle Important for Treatment?
Antiretroviral drugs target specific stages: entry inhibitors block binding/fusion, nucleoside reverse transcriptase inhibitors halt reverse transcription, integrase inhibitors prevent genome insertion, protease inhibitors stop maturation, and budding/maturation inhibitors are in development.
How Antiretroviral Drugs Target Specific Stages of the HIV Lifecycle
Antiretroviral therapy (ART) uses multiple drug classes, each targeting a specific stage of the HIV life cycle to suppress viral replication effectively.
How Do Entry and Fusion Inhibitors Block HIV?
Entry inhibitors like CCR5 antagonists (e.g., maraviroc) block HIV’s gp120 from binding to CCR5 co-receptors on CD4 cells; post-attachment inhibitors prevent co-receptor engagement. Fusion inhibitors such as enfuvirtide stop the viral envelope from merging with the cell membrane.
How Do RTIs Block Reverse Transcription?
Nucleoside reverse transcriptase inhibitors (NRTIs, e.g., lamivudine, emtricitabine) mimic nucleotides to halt RNA to DNA conversion; non-nucleoside reverse transcriptase inhibitors (NNRTIs) directly disable the reverse transcriptase enzyme.
How Do Drugs Target Integration and Maturation?
Integrase strand transfer inhibitors (INSTIs) block integrase from inserting viral DNA into the host genome. Protease inhibitors prevent cleavage of viral polyproteins, yielding immature, non-infectious virions.
What is the Best Treatment Strategy for Suppressing HIV?
HAART combines at least two classes (often three drugs) for synergy, reducing resistance risk and making HIV undetectable. Newer capsid inhibitors like lenacapavir target multiple steps, including nuclear entry and maturation.
How Drug Resistance Develops in HIV Treatment
HIV drug resistance develops primarily due to the virus’s high mutation rate during reverse transcription, combined with selective pressure from antiretroviral therapy (ART), allowing mutant strains to replicate while sensitive ones are suppressed.
What Mechanisms Drive HIV Drug Resistance?
Poor adherence
Missing doses allows viral replication during subtherapeutic drug levels, amplifying mutants; adherence below 95% doubles resistance risk.
High viral load
More replication cycles increase mutation opportunities.
Prior treatment exposure
Sequential monotherapy rapidly selects resistance, unlike modern multidrug regimens with high genetic barriers (e.g., dolutegravir, requiring multiple mutations).
Transmitted resistance
Drug-resistant strains from untreated individuals with virologic failure can infect others.
Consequences and Management
Resistance leads to treatment failure as viral load rebounds and CD4 counts drop; genotypic testing identifies mutations to guide regimen switches. Strategies like boosted regimens and adherence support minimize emergence, with newer drugs like lenacapavir showing higher barriers.
What is the Structural Morphology of the HIV Virus?
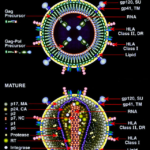
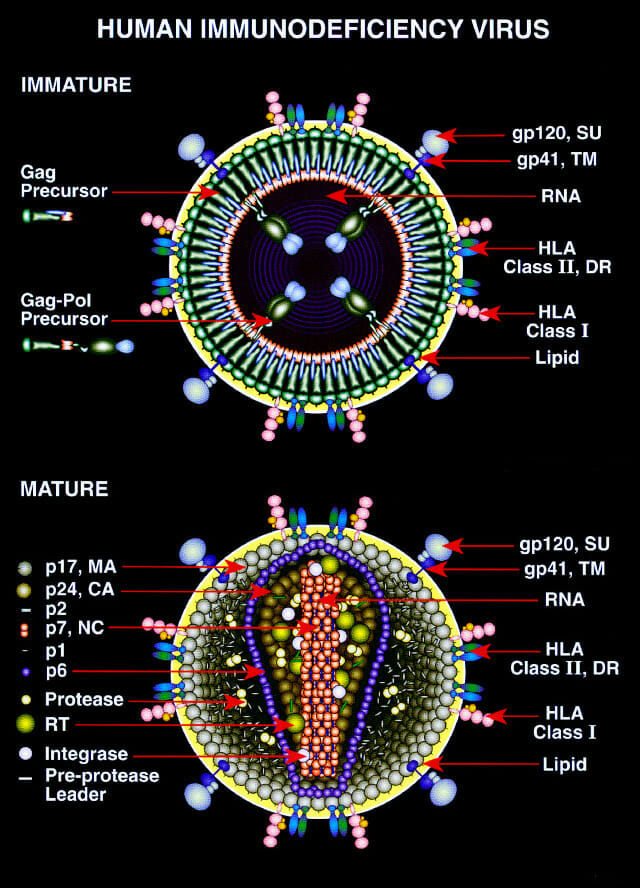
Human immunodeficiency virus (HIV) is a spherical, enveloped retrovirus about 100 to 120 nm in diameter, with a characteristic “doughnut-like” appearance in immature forms and a mature cone-shaped core inside the lipid envelope. Structurally, it follows the general organization of lentiviruses within the Retroviridae family and is composed of several distinct layers and components.
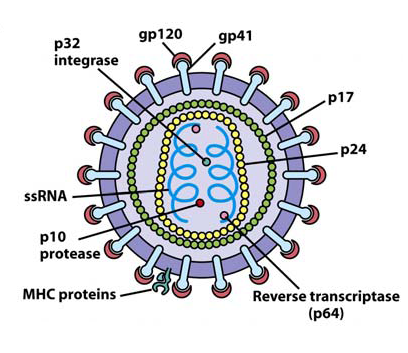
What is the Shape and Size of an HIV Virion?
HIV virions are roughly spherical and measure approximately 100 to 120 nm in diameter, making them much smaller than a red blood cell or typical host cell.
The outer surface shows irregular, knob-like spikes due to protruding viral glycoproteins, giving the virus a somewhat pleomorphic appearance under electron microscopy.
Envelope and Glycoproteins
The outer surface is a lipid bilayer derived from the host-cell plasma membrane, studded with viral envelope glycoproteins.
The envelope spikes are trimers of gp120 (surface glycoprotein) noncovalently associated with gp41 (transmembrane glycoprotein), which mediate attachment to CD4 and coreceptors on target cells.
Matrix and Capsid Core
Beneath the envelope lies a matrix layer made of the viral protein p17 (MA), which links the envelope to the internal core.
The interior contains a conical capsid composed mainly of p24 (CA) protein, enclosing the viral RNA genome and associated enzymes.
Genome and Internal Components
The core houses two identical, non-covalently linked copies of positive-sense, single-stranded RNA (about 9 to 10 kb) plus the viral enzymes reverse transcriptase, integrase, and protease.
The RNA is complexed with the nucleocapsid protein p7 (NC) and accessory proteins such as Vpr and others, forming a ribonucleoprotein complex inside the capsid.
FAQ
Which enzyme inserts HIV DNA into the host genome?
The integrase enzyme is responsible for inserting the viral DNA into the host cell’s genome during the integration phase.
Why does HIV develop drug resistance so quickly?
HIV has an exceptionally high mutation rate during reverse transcription and replicates rapidly, allowing mutant strains to survive when selective pressure from irregular ART is present.